Single-Cell Omics (SCO)
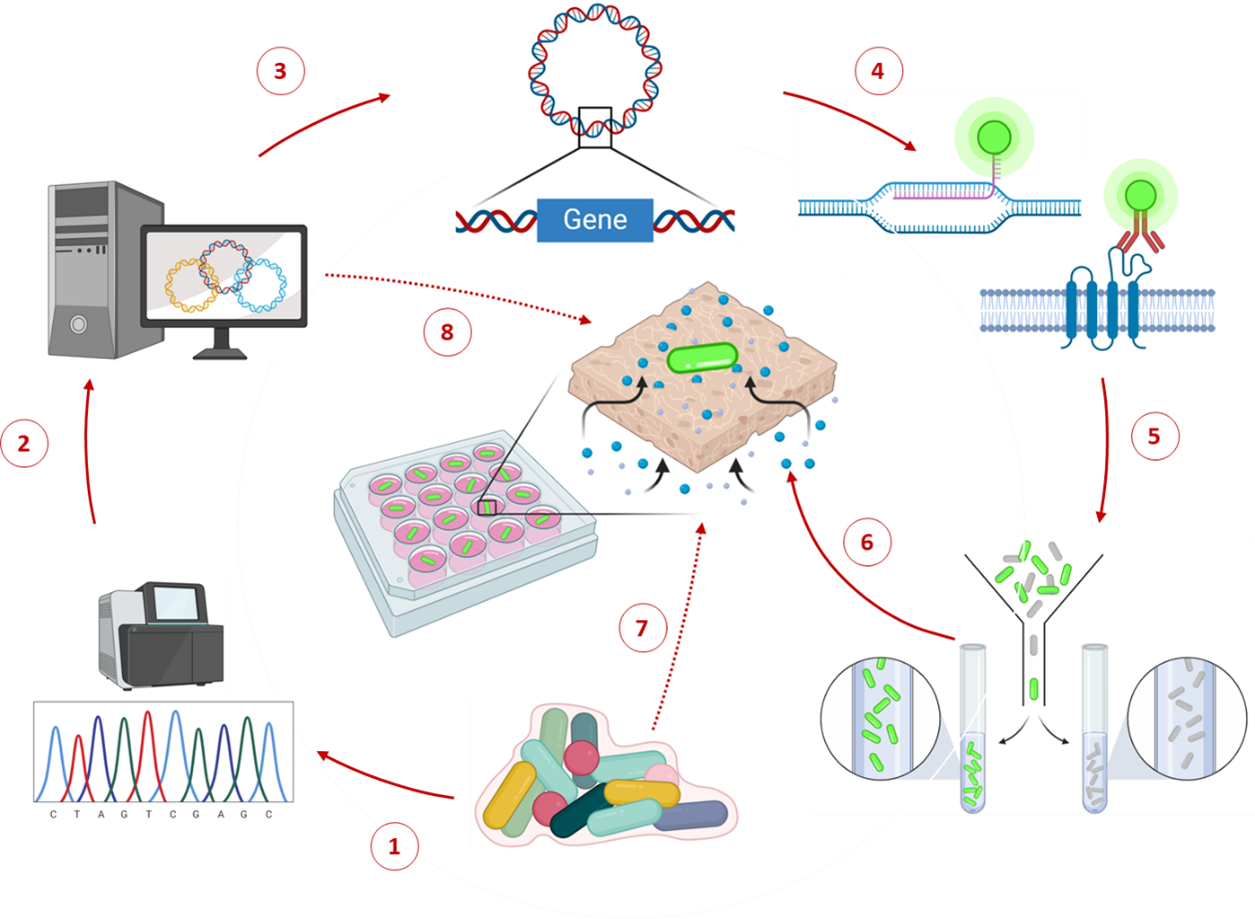
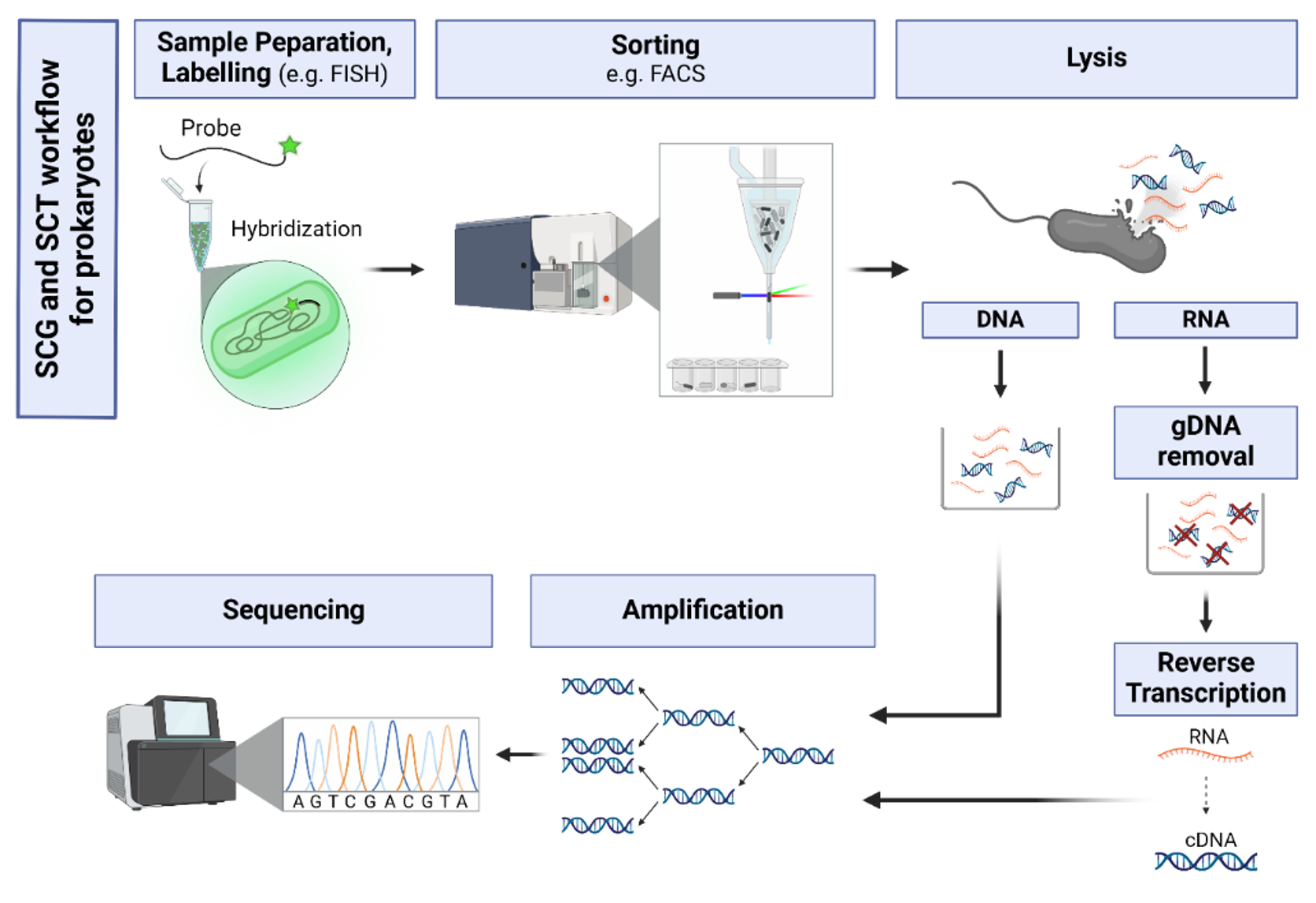
Single-cell omics has enabled us to better understand the diversity and functional heterogeneity of complex microbial communities, which is otherwise difficult with meta-omics methods. However, this field of research is still relatively new and there remain many methodological challenges that make it difficult to analyze single cells, especially from environmental samples. Our goal is to improve on currently existing methods in single cell genomics (SCG) and transcriptomics (SCT) so that these methods are more feasible for researchers to use and encourages the discovery of novel, uncultured prokaryotes (microbial dark matter).
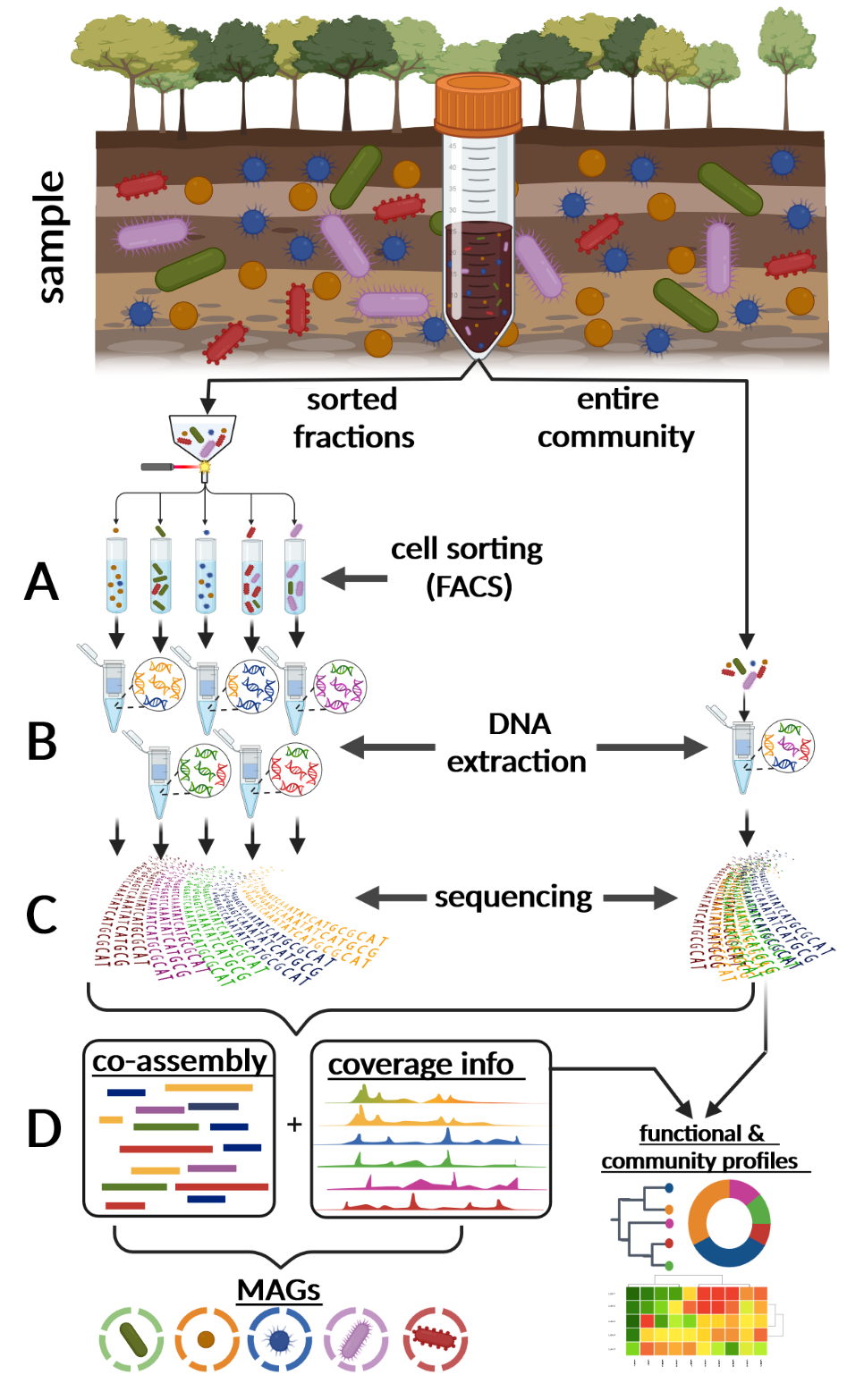
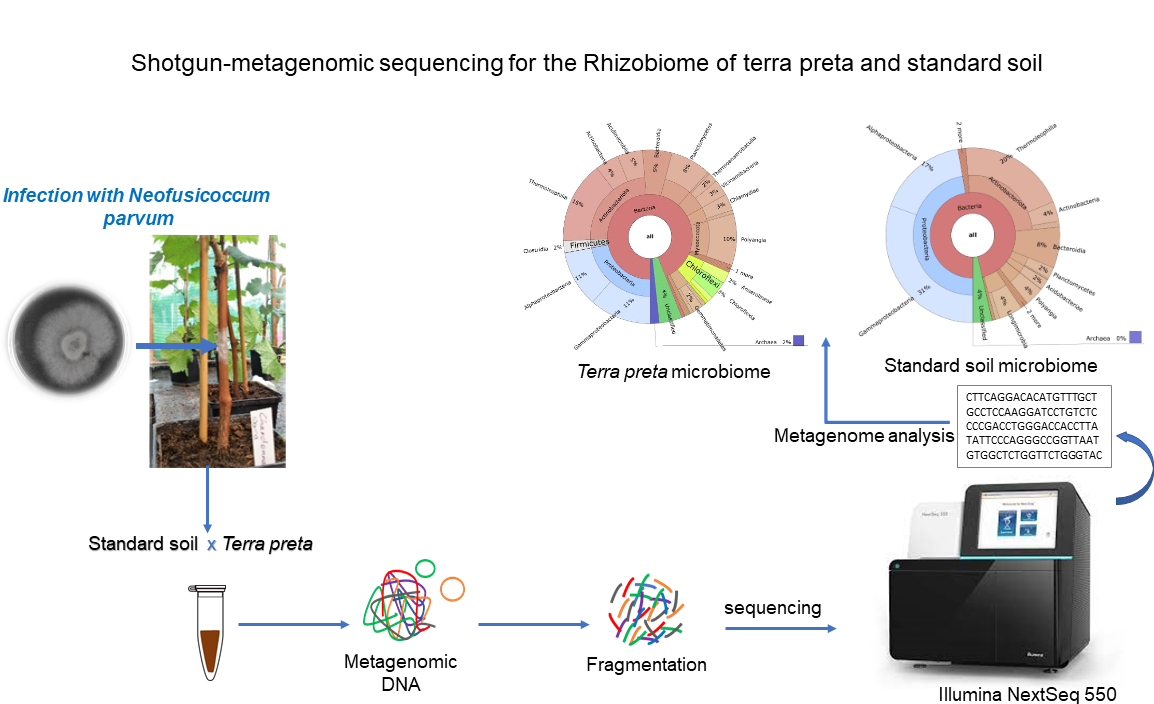
Integration and binning of single cell omics (SCO) with environmental data provide unprecedented insights into microbial diversity and metabolic features.
We currently host the first prokaryotic SCO pipeline in Europe, that
- enables the targeted selection of rare taxa for a deep investigation of microbial dark matter
- allows to sort prokaryotic cells under anoxic conditions
- improves cost-efficiency